Tidy Verbs for Dealing with Genomic Data Frames
Description
Handle genomic data within data frames just as you would with GRanges. This packages provides method to deal with genomics intervals the “tidy-way” which makes it simpler to integrate in the the general data munging process. The API is inspired by the popular bedtools and the genome_join() method from the fuzzyjoin package.
Installation
install.packages("tidygenomics")
# Or to get the latest development version
devtools::install_github("const-ae/tidygenomics")Documentation
genome_intersect
Joins 2 data frames based on their genomic overlap. Unlike the genome_join function it updates the boundaries to reflect the overlap of the regions.
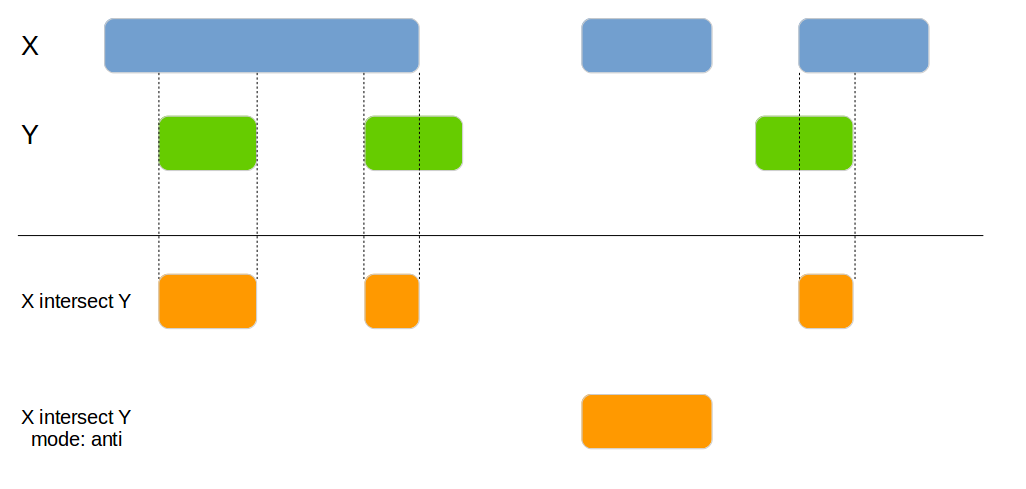
x1 <- data.frame(id = 1:4,
chromosome = c("chr1", "chr1", "chr2", "chr2"),
start = c(100, 200, 300, 400),
end = c(150, 250, 350, 450))
x2 <- data.frame(id = 1:4,
chromosome = c("chr1", "chr2", "chr2", "chr1"),
start = c(140, 210, 400, 300),
end = c(160, 240, 415, 320))
genome_intersect(x1, x2, by=c("chromosome", "start", "end"), mode="both")| id.x | chromosome | id.y | start | end |
|---|---|---|---|---|
| 1 | chr1 | 1 | 140 | 150 |
| 4 | chr2 | 3 | 400 | 415 |
genome_subtract
Subtracts one data frame from the other. This can be used to split the x data frame into smaller areas.
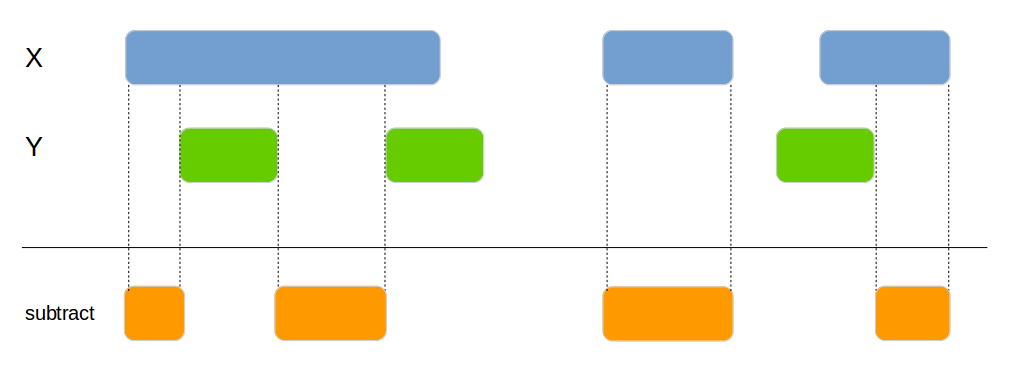
x1 <- data.frame(id = 1:4,
chromosome = c("chr1", "chr1", "chr2", "chr1"),
start = c(100, 200, 300, 400),
end = c(150, 250, 350, 450))
x2 <- data.frame(id = 1:4,
chromosome = c("chr1", "chr2", "chr1", "chr1"),
start = c(120, 210, 300, 400),
end = c(125, 240, 320, 415))
genome_subtract(x1, x2, by=c("chromosome", "start", "end"))| id | chromosome | start | end |
|---|---|---|---|
| 1 | chr1 | 100 | 119 |
| 1 | chr1 | 126 | 150 |
| 2 | chr1 | 200 | 250 |
| 3 | chr2 | 300 | 350 |
| 4 | chr1 | 416 | 450 |
genome_join_closest
Joins 2 data frames based on their genomic location. If no exact overlap is found the next closest interval is used.
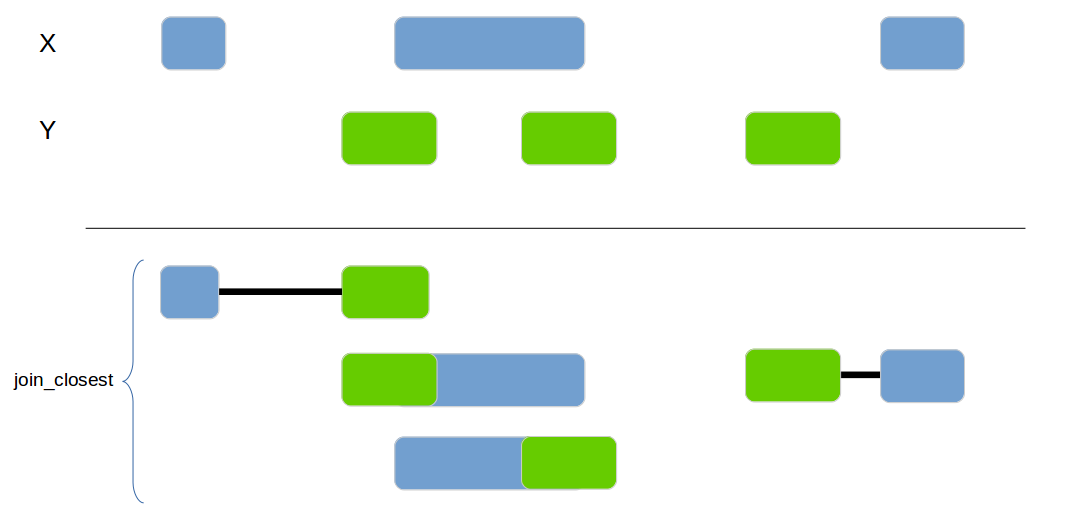
x1 <- data_frame(id = 1:4,
chr = c("chr1", "chr1", "chr2", "chr3"),
start = c(100, 200, 300, 400),
end = c(150, 250, 350, 450))
x2 <- data_frame(id = 1:4,
chr = c("chr1", "chr1", "chr1", "chr2"),
start = c(220, 210, 300, 400),
end = c(225, 240, 320, 415))
genome_join_closest(x1, x2, by=c("chr", "start", "end"), distance_column_name="distance", mode="left")| id.x | chr.x | start.x | end.x | id.y | chr.y | start.y | end.y | distance |
|---|---|---|---|---|---|---|---|---|
| 1 | chr1 | 100 | 150 | 2 | chr1 | 210 | 240 | 59 |
| 2 | chr1 | 200 | 250 | 1 | chr1 | 220 | 225 | 0 |
| 2 | chr1 | 200 | 250 | 2 | chr1 | 210 | 240 | 0 |
| 3 | chr2 | 300 | 350 | 4 | chr2 | 400 | 415 | 49 |
| 4 | chr3 | 400 | 450 | NA | NA | NA | NA | NA |
genome_cluster
Add a new column with the cluster if 2 intervals are overlapping or are within the max_distance.
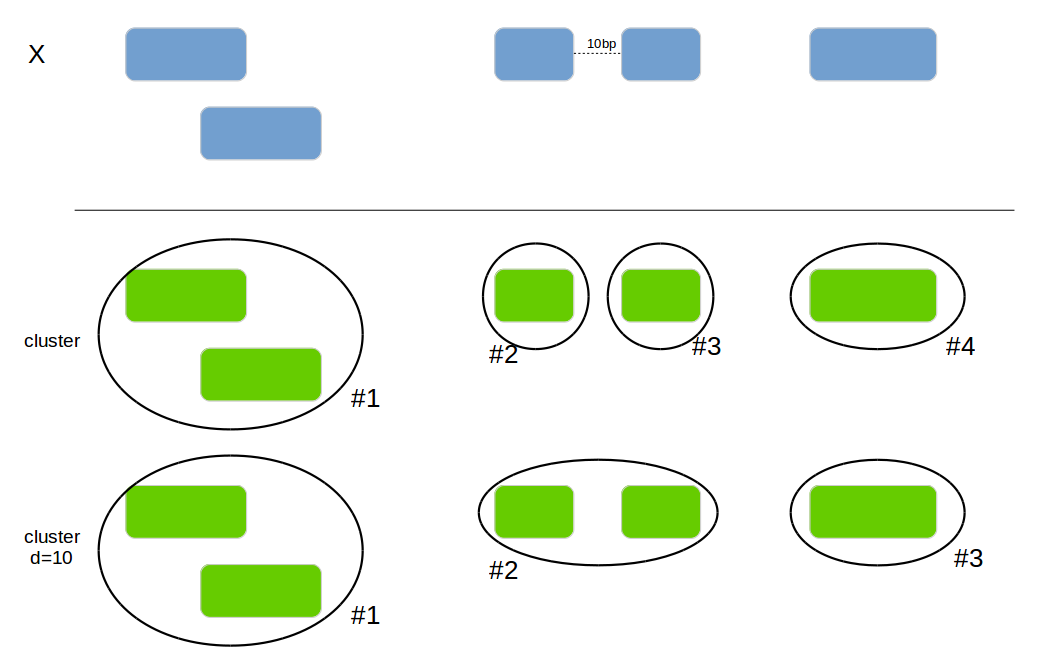
x1 <- data.frame(id = 1:4, bla=letters[1:4],
chromosome = c("chr1", "chr1", "chr2", "chr1"),
start = c(100, 120, 300, 260),
end = c(150, 250, 350, 450))
genome_cluster(x1, by=c("chromosome", "start", "end"))| id | bla | chromosome | start | end | cluster_id |
|---|---|---|---|---|---|
| 1 | a | chr1 | 100 | 150 | 0 |
| 2 | b | chr1 | 120 | 250 | 0 |
| 3 | c | chr2 | 300 | 350 | 2 |
| 4 | d | chr1 | 260 | 450 | 1 |
genome_cluster(x1, by=c("chromosome", "start", "end"), max_distance=10)| id | bla | chromosome | start | end | cluster_id |
|---|---|---|---|---|---|
| 1 | a | chr1 | 100 | 150 | 0 |
| 2 | b | chr1 | 120 | 250 | 0 |
| 3 | c | chr2 | 300 | 350 | 1 |
| 4 | d | chr1 | 260 | 450 | 0 |
genome_complement
Calculates the complement of a genomic region.
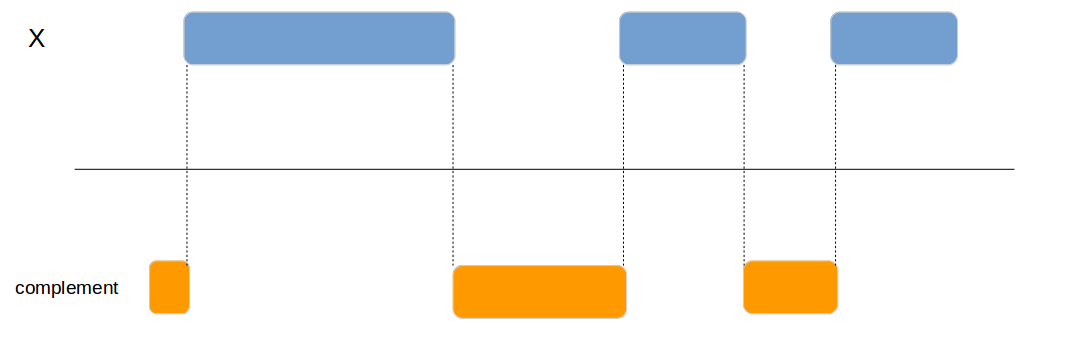
x1 <- data.frame(id = 1:4,
chromosome = c("chr1", "chr1", "chr2", "chr1"),
start = c(100, 200, 300, 400),
end = c(150, 250, 350, 450))
genome_complement(x1, by=c("chromosome", "start", "end"))| chromosome | start | end |
|---|---|---|
| chr1 | 1 | 99 |
| chr1 | 151 | 199 |
| chr1 | 251 | 399 |
| chr2 | 1 | 299 |
genome_join
Classical join function based on the overlap of the interval. Implemented and maintained in the fuzzyjoin package and documented here only for completeness.

x1 <- data_frame(id = 1:4,
chr = c("chr1", "chr1", "chr2", "chr3"),
start = c(100, 200, 300, 400),
end = c(150, 250, 350, 450))
x2 <- data_frame(id = 1:4,
chr = c("chr1", "chr1", "chr1", "chr2"),
start = c(220, 210, 300, 400),
end = c(225, 240, 320, 415))
fuzzyjoin::genome_join(x1, x2, by=c("chr", "start", "end"), mode="inner")| id.x | chr.x | start.x | end.x | id.y | chr.y | start.y | end.y |
|---|---|---|---|---|---|---|---|
| 2 | chr1 | 200 | 250 | 1 | chr1 | 220 | 225 |
| 2 | chr1 | 200 | 250 | 2 | chr1 | 210 | 240 |
fuzzyjoin::genome_join(x1, x2, by=c("chr", "start", "end"), mode="left")| id.x | chr.x | start.x | end.x | id.y | chr.y | start.y | end.y |
|---|---|---|---|---|---|---|---|
| 1 | chr1 | 100 | 150 | NA | NA | NA | NA |
| 2 | chr1 | 200 | 250 | 1 | chr1 | 220 | 225 |
| 2 | chr1 | 200 | 250 | 2 | chr1 | 210 | 240 |
| 3 | chr2 | 300 | 350 | NA | NA | NA | NA |
| 4 | chr3 | 400 | 450 | NA | NA | NA | NA |
fuzzyjoin::genome_join(x1, x2, by=c("chr", "start", "end"), mode="anti")| id | chr | start | end |
|---|---|---|---|
| 1 | chr1 | 100 | 150 |
| 3 | chr2 | 300 | 350 |
| 4 | chr3 | 400 | 450 |
Inspiration
If you have any additional questions or encounter issues please raise them on the github page.